

Struttura del glicogeno, sintesi, degradazione, funzioni
- 2724
- 91
- Lino Lombardi
Lui Glicogeno È il carboidrato di stoccaggio della maggior parte dei mammiferi. I carboidrati sono comunemente chiamati zuccheri e questi sono classificati in base al numero di rifiuti causati dall'idrolisi (monosaccaridi, disaccaridi, oligosaccaridi e polisaccaridi)).
I monosaccaridi sono i carboidrati più semplici che sono classificati in base al numero di carboni contenuti nella loro struttura. Ci sono quindi le Triose (3C), Tetrosas (4C), Pentosas (5C), esoso (6C), Eptosase (7C) e Octosas (8C).
Struttura del glicogeno chimico che mostra legami glicosidici (fonte: Glykogen.SVG: Neurotoger Derivative Work: Marek M [dominio pubblico] via Wikimedia Commons) Secondo la presenza del gruppo aldeide o del gruppo Cetona, questi monosaccaridi sono anche classificati rispettivamente come aldies o ketosas.
I disaccaridi danno origine all'idrolisi, due semplici monosaccaridi, mentre gli oligosaccaridi producono da 2 a 10 unità di monosaccaridi e polisaccaridi producono più di 10 monosaccaridi.
Il glicogeno è, dal punto di vista biochimico, un polisaccaride composto da catene ramificate di un aldosio a sei carbonio, cioè un esoso noto come glucosio. Graficamente può essere rappresentato a glicogeno come un albero di glucosio. Questo è anche chiamato amido animale.
Il glucosio nelle piante è immagazzinato come amido e negli animali come glicogeno, che è conservato principalmente nel fegato e nel tessuto muscolare.
Nel fegato, il glicogeno può stabilire il 10% della sua massa e l'1% della massa muscolare. Come in un uomo di 70 kg, il fegato pesa circa 1800 g e i muscoli di circa 35 kg, la quantità totale di glicogeno muscolare è molto maggiore dell'epatico.
[TOC]
Struttura
Il peso molecolare del glicogeno può raggiungere 108 g/mol, equivalente a molecole di glucosio 6 × 105. Il glicogeno è costituito da catene α-D-glicosio ramificate multiple. Il glucosio (C6H12O6) è un Aldohexosa che può essere rappresentato in modo lineare o ciclico.
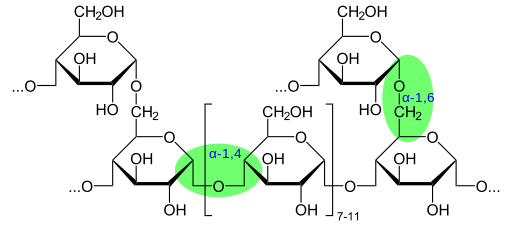
Il glicogeno ha una struttura molto ramificata e compatta con catene da 12 a 14 rifiuti di glucosio sotto forma di α-D-glucosio che sono collegati a legami glucosidici α- (1 → 4). Le ramificazioni a catena sono formate da collegamenti glucosidici α- (1 → 6).
Il glicogeno, come l'amido che viene ingerito nella dieta, fornisce la maggior parte dei carboidrati di cui il corpo ha bisogno. Nell'intestino questi polisaccaridi sono degradati dall'idrolisi e quindi assorbiti verso il torrente circolatorio principalmente come glucosio.
Tre enzimi: ß-amilasi, α-amilasi e amilo-α- (1 → 6) -glucosidasi sono responsabili della degradazione intestinale sia del glicogeno che dell'amido.
L'α-amilasi idrolizza casualmente i legami α (1 → 4) delle catene laterali sia del glicogeno che dell'amido, e quindi riceve il nome dell'endoglysidasi. La ß-amyla è un'esoglicosidasi che sta rilasciando i collegamenti glicosidici α- (1 → 4) che rompono le estremità delle catene più esterne senza raggiungere le ramificazioni.
In considerazione del fatto che né ß-amilasi né α-amilasi degradano i rami, il prodotto finale della sua azione è una struttura altamente ramificata di circa 35 a 40 residui di glucosio che sono chiamati il limite di destrina.
La destrina limite viene infine idrolizzata nei punti di ramo che hanno α- (1 → 6) si legano attraverso l'amyle-α- (1 → 6) -glucosidasi, noto anche come enzima "diffamatorio". Le catene rilasciate da questo defloat sono dopo degradate da ß-amilasi e α-amilasi.
Poiché il glicogeno ingerito entra come glucosio, quello trovato nei tessuti deve essere sintetizzato dall'organismo dal glucosio.
Può servirti: purine: caratteristiche, struttura, funzioniSintesi
La sintesi del glicogeno si chiama glicogenesi e si svolge soprattutto nel muscolo e nel fegato. Il glucosio che entra nell'organismo con la dieta passa al torrente circolatorio e da lì all'interno delle cellule, dove viene immediatamente fosforilato da un enzima chiamato glycoquinasi.
Glucochinasi fosforil al glucosio in carbonio 6. L'ATP fornisce fosforo e energia per questa reazione. Di conseguenza, si forma il glucosio 6-fosfato e viene rilasciato un ADP. Quindi, il glucosio a 6-fosfato diventa glucosio 1-fosfato mediante l'azione di una fosfoglucomutasi che infrange il fosforo dalla posizione 6 alla posizione 1.
Il glucosio 1-fosfato è attivato per la sintesi del glicogeno, che implica la partecipazione di un insieme di altri tre enzimi: la pirofosforilasi UDP-glicosa, il glicogeno sintetico e l'amilo- (1.4 → 1.6) -glicosiltransferasi.
Il glucosio-1-fosfato, insieme al trifosfato uridina (UTP, un nucleoside di uridina trifosfato) e mediante azione dell'UDP-glicosio-pirofosforilasi, formano il complesso di uridina di difosfato-glucosio (UDP GLC) (UDP GLC). Nel processo viene idrolizzato uno ione pirofosfato.
Quindi, l'enzima glicogeno sintetico forma un legame glucosidico tra il C1 del complesso GLC UDP e il C4 di un residuo di glicogeno di glucosio e l'UDP UDP viene rilasciato dal complesso di glucosio attivato da UDP. Perché questa reazione si verifichi, deve esserci una molecola di glicogeno pre -esistente chiamata "glicogeno primario".
Il glicogeno primordiale è sintetizzato su una proteina di innesco, glicogenina, che ha 37 kDa e glysila in un residuo di tirosina usando il complesso UDP GLC. Da lì sono collegati rifiuti α-D-glucosio con collegamenti 1 → 4 e si forma una piccola catena su cui agisce il glicogeno Syntesasi.
Una volta che la catena iniziale collega almeno 11 residui di glucosio, la ramificazione o l'enzima amile (1,4 → 1,6) -glicosiltransferasi trasferisce un pezzo a catena di 6 o 7 rifiuti di glucosio nella catena adiacente in posizione 1 → 6, che stabilisce un ramo punto. La molecola di glicogeno così costruita sta crescendo con aggiunte di unità di glucosio con collegamenti glicosidici 1 → 4 e più ramificazioni.
Degradazione
La degradazione del glicogeno è chiamata glucogenolisi e non è equivalente al percorso inverso della sua sintesi. La velocità di questo percorso è limitata dalla velocità della reazione catalizzata dal glicogeno fosforilasi.
Il glicogeno della fosforillasi è responsabile della divisione (fosforolisi) dei collegamenti 1 → 4 dalle catene di glicogeno, rilasciando glucosio 1-fosfato. L'azione enzimatica inizia alle estremità delle catene più esterne e vengono rimosse in sequenza fino a quando 4 residui di glucosio rimangono su ciascun lato delle ramificazioni.
Quindi, un altro enzima, le transferes di glucano α- (1 → 4) → α- (1 → 4), lascia il punto di ramo esposto trasferendo un'unità di trisaccaride da un ramo a un altro. Ciò consente l'amilo- (1 → 6) -glucosidasi (enzima non rempico) idrolys. L'azione combinata di questi enzimi termina completamente di divisione in glicogeno.
Poiché la reazione iniziale della fosfomutasi è reversibile, il glucosio a 6-fosfato può essere formato da residui di glucosio 1-fosfato diviso dal glicogeno. Nel fegato e nel rene, ma non nel muscolo, c'è un enzima, glucosio-6-fosfatasi, in grado di raccogliere al glucosio a 6 fosfato e trasformarlo in glucosio libero.
Può servirti: fotolisiIl glucosio defosforilato può diffondersi al sangue, ed è così che la glicogenolisi epatica si riflette in un aumento dei valori del glucosio nel sangue (glicemia).
Regolazione della sintesi e del degrado
Di sintesi
Questo processo è esercitato su due enzimi fondamentali: glicogeno sintesi e glicogeno fosforilasi, in modo che quando uno di essi è attivato, l'altro è nel suo stato inattivo. Questa regolazione impedisce le reazioni opposte di sintesi e degradazione dal verificarsi simultaneamente.
La forma attiva e la forma inattiva di entrambi gli enzimi sono molto diverse e l'interconversione delle forme attive e inattive di fosforilasi e glicogeno sintetico è soggetta a un rigoroso controllo ormonale.
L'adrenalina è un ormone che viene rilasciato dal midollo surrenale e il glucagone è un altro che si verifica nella parte endocrina del pancreas. Il pancreas endocrino produce insulina e glucagone. Le isole di Langerhans α sono quelle che sintetizzano il glucagone.
L'adrenalina e il glucagone sono due ormoni che vengono rilasciati quando è necessaria energia in risposta alla diminuzione dei livelli di glucosio nel sangue. Questi ormoni stimolano l'attivazione del glicogeno della fosforilasi e inibiscono il glicogeno della sintesi, stimolando così la glicogenolisi e inibendo la glicogenesi.
Mentre l'adrenalina esercita la sua azione sul muscolo e sul fegato, il glucagone agisce solo sul fegato. Questi ormoni sono uniti a specifici recettori membranali nei globuli bianchi, che attiva CyclASA adeniato.
L'attivazione della ciclasi adenilata inizia una cascata enzimatica che, da un lato, attiva una proteinquinasi dipendente dall'AMPC che inattiva al glicogeno sintetico e attiva la fosforilasi glicogeno mediante fosforilazione (direttamente e indirettamente, rispettivamente).
Il muscolo scheletrico ha un altro meccanismo per l'attivazione del glicogeno fosforilasi attraverso il calcio, che viene rilasciato a seguito della depolarizzazione della membrana muscolare all'inizio della contrazione.
Di degrado
Le cascate enzimatiche descritte sopra finiscono per aumentare i livelli di glucosio e quando raggiungono un certo livello, la glicogenesi viene attivata e la glucogenolisi è inibita, inibendo anche l'ulteriore rilascio di adrenalina e glucagone.
La glicogenesi è attivata dall'attivazione della fosfatasi fosforilasi, un enzima che regola la sintesi del glicogeno da parte di diversi meccanismi, che implicano l'inattivazione della chinasi fosforilasi e la fosforilasi α, che è un inibitore del glicogeno sintesi.
L'insulina promuove l'ingresso del glucosio nelle cellule muscolari, aumentando i livelli di glucosio a 6-fosfato, che stimola la defosforilazione e l'attivazione del glicogeno sintesi. Pertanto inizia la sintesi e la degradazione del glicogeno è inibita.
Funzioni
Il glicogeno muscolare costituisce una riserva energetica per il muscolo che, come i grassi di riserva, consente ai muscoli di svolgere le sue funzioni. Essendo una fonte di glucosio, il glicogeno muscolare viene utilizzato durante l'esercizio. Queste riserve aumentano con l'allenamento fisico.
Nel fegato, il glicogeno costituisce anche un'importante fonte di riserva sia per le funzioni dell'organo sia per il contributo del glucosio al resto del corpo.
Questa funzione del glicogeno epatico è dovuta al fatto che il fegato contiene glucosio 6-fosfatasi, un enzima in grado di eliminare il gruppo fosfato di glucosio a 6 fosfato e trasformarlo in glucosio libero. Il glucosio libero, a differenza del glucosio fosforilato, può essere diffuso attraverso la membrana degli epatociti (cellule epatiche).
Può servirti: sporulazione: nelle piante, nei funghi e nei batteriEcco come il fegato può fornire glucosio alla circolazione e mantenere livelli stabili di glucosio, anche in condizioni di digiuno prolungate.
Questa funzione è di grande importanza, poiché il cervello è nutrito quasi esclusivamente dalla glicemia, quindi un'ipoglicemia grave (concentrazioni di glucosio nel sangue molto basse) può causare perdita di conoscenza.
Malattie correlate
Le malattie correlate al glicogeno ricevono il nome generico di "Malattie di conservazione del glicogeno".
Queste malattie costituiscono un gruppo di patologie ereditarie caratterizzate dal deposito nei tessuti di quantità anormali o tipi di glicogeno.
La maggior parte delle malattie di stoccaggio del glicogeno sono causate da un deficit naturale genetico di uno qualsiasi degli enzimi coinvolti nel metabolismo del glicogeno.
Sono classificati in otto tipi, la maggior parte dei quali ha i propri nomi e ognuno di essi è prodotto da un diverso deficit enzimatico. Alcuni sono mortali nelle prime fasi della vita, mentre altri sono accompagnati da debolezza muscolare e deficit durante l'esercizio fisico.
Esempi eccezionali
Alcune delle più importanti malattie correlate al glicogeno sono le seguenti:
- La malattia di Von Gierke o la malattia di stoccaggio del glicogeno di tipo I, è prodotta da un deficit di glucosio 6-fosfatasi nel fegato e nel rene.
È caratterizzato da una crescita epatica anormale (epatomegalia) a causa dell'accumulo esagerato di glicogeno e ipoglicemia, poiché il fegato non è in grado di fornire glucosio alla circolazione. I pazienti con questa condizione hanno alterazioni di crescita.
- Pompe o malattia di tipo II è dovuta a un deficit α-deficit (1 → 4) -Glucano 6-glicosiltransferas nel fegato, nel cuore e nei muscoli scheletrici. Questa malattia, come Andersen o il tipo IV, è letale prima dei due anni di vita.
- La malattia di McArdle o di tipo V ha un deficit di fosforilasi muscolare ed è accompagnata da debolezza muscolare, ridotta tolleranza all'esercizio fisico, accumulo anormale di glicogeno muscolare e assenza di lattato durante l'esercizio fisico.
Riferimenti
- Bhattacharya, k. (2015). Indagine e gestione delle malattie epatiche di stoccaggio del glicogeno. Pediatria traslazionale, 4(3), 240-248.
- Dagli, a., Sendner, c., & Weinstein, D. (2016). Disegno di stoccaggio del glicogeno Tipo III. Recensioni geniche, 1-16.
- Guyton, a., & Hall, J. (2006). Libro di testo di fisiologia medica (11 ° ed.). Elsevier Inc.
- Mathews, c., Van Holde, K., & Ahern, K. (2000). Biochimica (3 ° ed.). San Francisco, California: Pearson.
- McKiernan, p. (2017). Patobiologia del desiderio epatico di stoccaggio del glicogeno. Curr Pathobiol Rep.
- Murray, r., Bender, d., Botham, k., Kennelly, p., Rodwell, v., & Weil, p. (2009). Biochimica illustrata di Harper (28 ° ed.). McGraw-Hill Medical.
- Nelson, d. L., & Cox, m. M. (2009). Principi di biochimica Lechinger. Omega Editions (5 ° ed.).
- Rawn, j. D. (1998). Biochimica. Burlington, Massachusetts: Neil Patterson Publishers.
- Tarnopolsky, m. A. (2018). Miopatie correlate ai disturbi del metabolismo del glicogeno. Neuroterapici.
- « Storia, struttura, proprietà, usi dell'argon
- Funzione di bijective Cos'è, come è fatto, esempi, esercizi »