

Prioni
- 3910
- 670
- Ruth Cattaneo
I prioni sono proteine scarsamente piegate che trasmettono queste informazioni errate ad altre proteine e causano encefalopatie spongiformi trasmissibili. Fonte: Wikimedia Commons Cosa sono i prioni?
IL Prioni Sono proteine senza genoma o acidi nucleici che agiscono come agenti infettivi. Si trovano nella normale membrana cellulare, solo come proteine scarsamente piegate e/o con struttura tridimensionale anormale.
Queste proteine sono responsabili di malattie degenerative multiple e mortalità molto elevata che influenzano i tessuti neurali e la struttura cerebrale.
Sono anche chiamati malattie prioniche. Tra i più importanti che colpiscono gli umani ci sono Kuru, la malattia di Gerstmann-Sträussler-Scheinker, la sindrome di Creutzfeldt-Jakob e l'insonnia familiare fatale.
Caratteristiche del prione
- I proni sono strutture proteiche presenti nelle membrane cellulari. Queste proteine hanno una forma alterata o una conformazione [PRP (SC)].
- Per quanto riguarda la sua moltiplicazione, si ottiene mediante la conversione delle forme, come nel caso della malattia tremante. In questa malattia, i prioni reclutano PRP (c) (proteine prionali della conformazione incustodita) per stimolare la conversione all'isoforma del PRP (SC).
- Queste insolite proteine in grado di diffondersi, non hanno acidi nucleici. La prova di ciò è che sono resistenti ai raggi X e alle radiazioni ultraviolette. Questi agenti abbattono facilmente gli acidi nucleici.
- Le proteine prioniche, di cui sono composte i prioni (PRP), si trovano in tutto il corpo, non solo degli esseri umani ma di altri vertebrati sani.
- Alcuni ricercatori sono riusciti a dimostrare che, nei topi, queste proteine attivano la riparazione mielinica nelle cellule del sistema nervoso periferico. È stato anche dimostrato che l'assenza di questi provoca la demielinizzazione di tali cellule nervose.
Struttura del prione
La conoscenza della struttura dei prioni risiede principalmente nelle indagini condotte nei batteri Escherichia coli.
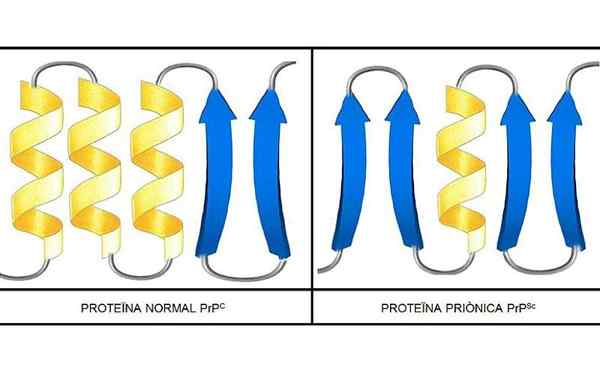
Gli studi hanno condotto che i polipeptidi nella catena PRP (c) (normale) e PRP (SC) (infettivo) sono identici nella composizione degli aminoacidi, ma differiscono nella conformazione 3D e nella piegatura di questi.
PRP (C)
Questi prioni non infettivi presenti, nell'uomo, 209 aminoacidi. Hanno un collegamento disolfuro. La sua struttura è alfa-elicoidale, il che significa che ha aminoacidi a forma di spirale (eliche alfa) e pochi fili di aminoacidi piatti (foglie beta).
Può servirti: scienze ausiliarie della biologiaQuesta proteina non può essere separata dalla centrifugazione, il che implica che non è sedimentabile. Viene facilmente digerito dall'ampia proteasi di ampio spettro chiamato proteinasi K.
PRP (SC)
È una proteina infettiva che trasforma PRP (C) in isoforme PRP (SC) infettive.
Si sa molto poco sulla sua struttura 3D, tuttavia è noto che ha poche forme elicoidali e più fili piatti o foglie beta. Il cambiamento verso l'isoforma è ciò che è noto come l'evento fondamentale delle malattie del prione.
Funzioni di prione
Le proteine del prione di cellule [PRP (c)] si trovano sulla superficie cellulare di un'ampia varietà di organi e tessuti. Si sa molto poco sulle funzioni fisiologiche dei prioni nel corpo.
Anche così, le esperienze fatte nei topi indicano possibili funzioni, come:
Con recettori del glutammato metabotropico
È stato dimostrato che il PRP (C) agisce con i recettori del glutammato (ionotropici e metabotropici). Il PRP (c) partecipa come recettore degli oligomeri sinaptotossici del peptide cellulare della superficie cellulare.
Nello sviluppo embrionale
Nei topi familiari Murinae, è stato scoperto che le proteine del PRP (C) Prion sono espresse pochi giorni dopo l'impianto, nello sviluppo embrionale.
Ciò indica che svolgono un ruolo durante lo sviluppo di questi piccoli mammiferi. Documento che secondo i ricercatori è correlato alla regolazione della neuritogenesi (produzione di assoni e dendriti dei neuroni).
Agiscono anche in crescita assonale. Queste proteine del prione sono persino coinvolte nello sviluppo del circuito cerebellare. Per questo motivo, si ritiene che l'assenza di questi prioni PRP (c) implica un ritardo nello sviluppo motorio dei roditori.
Neuroprotettore
Negli studi sulla sovraespressione di PRP (c) a causa dell'orientamento dei geni, è stato scoperto che l'assenza di questi prioni provoca problemi con l'irrigazione del sangue in alcuni luoghi del cervello (ischemia cerebrale acuta).
Ciò significa che le proteine del prione funzionano come neuroprotettori. Inoltre, è stato dimostrato che la sovraespressione di PRP (C) può ridurre o migliorare le lesioni causate dall'ischemia.
Sistema nervoso periferico
Recentemente, la funzione fisiologica di PRP (C) è stata scoperta nel mantenimento della mielina periferica.
Può servirti: distrofina: caratteristiche, struttura e funzioniDurante uno studio di laboratorio è stato scoperto che in assenza di proteine del prione, i topi di laboratorio hanno sviluppato carenze nervose che trasferiscono le informazioni dal cervello e dal midollo spinale, in quella che viene chiamata neuropatia periferica.
Morte cellulare
Ci sono alcune proteine prioniche e si trovano in altre parti del corpo diverse dal cervello.
Le funzioni di tali proteine sono di iniziare, regolare e/o controllare la morte cellulare, quando il corpo viene attaccato (ad esempio da vuloni), impedendo così la propagazione del patogeno.
Questa particolare funzione di queste proteine, fa pensare ai ricercatori alla possibile importanza dei prioni non infettivi nella lotta contro i patogeni.
Memoria a lungo termine
Uno studio condotto presso il Stowers Institute, nel Missouri, EE. Uu., Ha dimostrato che PRP Prions può avere una funzione nella manutenzione della memoria a lungo termine.
Lo studio ha rivelato che alcune proteine del prione possono essere controllate per lavorare sul mantenimento delle funzioni fisiologiche della memoria a lungo termine.
Rinnovo delle cellule madre
Un'indagine sulle proteine del prione che sono espresse nelle cellule del tessuto dello stelo, ha rivelato che tutte queste cellule staminali (ematopoietiche), esprimono proteine di prione nella loro membrana cellulare. Quindi si ritiene che partecipino al complesso e molto importante processo di rinnovamento delle cellule.
Malattie causate dai prioni
Le malattie del prione più comuni sono:
Malattia di Creutzfeldt-Jakob (ECJ) (ECJ)
Considerata la malattia del prione più comune tra gli esseri umani, è una patologia cosmopolita, cioè distribuzione mondiale. Può verificarsi ereditarietà (famiglia), sporadica o contagiosa.
Malattia di Gerstmann-Sträussler-Scheinker
È una malattia causata dai prioni in un processo encefalico dominante infettivo o autosomico ereditario. La malattia si manifesta in persone da 40 a 60 anni.
Prionopatia con sensibilità di proteasi variabile
È una malattia molto rara, al punto che la sua gamma di occorrenze è da 2 a 3 casi per 100 milioni di abitanti. La patologia è simile alla malattia di Gerstmann-Sträussler-Scheinker.
Insonnia letale
È una malattia ereditaria o familiare, sebbene possa verificarsi anche sporadicamente. È noto che la malattia è dovuta a una mutazione ereditaria o autosomica dominante.
Può servirti: specie endemicheKuru
Questa malattia del prione è stata rilevata solo negli abitanti della Papua Nuova Guinea. È una malattia legata al cannibalismo e alla tradizione culturale del rito del duello per i morti, dove queste persone mangiano il cervello umano.
Malattie negli animali
Tra le patologie prodotte da Prion negli animali vi è encefalopatia spongiforme bovina. Questa malattia ha provocato il caos in Europa, nella sanità pubblica, quella degli animali e nell'economia dei paesi colpiti.
Altre malattie negli animali sono scrapy, encefalopatia trasmissibile del visone, malattia da usura cronica (in cervo) e encefalopatia spongiforme felina.
Queste malattie, come quelle presentate nell'uomo, mancano di un trattamento efficace, quindi la prevenzione è fondamentale, specialmente dopo i contagios nell'uomo che si sono verificati a causa del consumo di carne di vacche infette.
Trattamenti
Ad oggi, non è nota nessuna cura per le malattie del prione. Il trattamento è sintomatico. Si consiglia ai pazienti di pianificare cure palliative e analisi genetiche e consigli per i familiari.
Un'ampia varietà di farmaci è stata testata in pazienti con malattie prioniche, come antivirale, antitumorale.
Tuttavia, al momento non ci sono prove che indicano che alcuni di questi diminuiscono i sintomi o migliorano la sopravvivenza dei malati.
Prevenzione
I proni sono resistenti a una varietà di cambiamenti fisici e chimici. Tuttavia, vengono utilizzate diverse tecniche per evitare l'inquinamento dei pazienti con strumenti chirurgici contaminati.
Tra le tecniche più comunemente utilizzate è sterilizzare l'attrezzatura in un'autoclave a 132 ° C per un'ora e quindi immergere gli strumenti in idrossido di sodio per almeno un'ora in più.
D'altra parte, l'Organizzazione mondiale della sanità (OMS) ha sviluppato misure per evitare la diffusione delle malattie del prione. Questa organizzazione stabilisce regole per la gestione di tessuti proibiti o potenzialmente rischiosi come: occhi, cervello, intestino, tonsille e midollo spinale.
Riferimenti
- Prione, agente contagioso. Recuperato dalla Britannica.com.
- Cos'è un prione? Recuperato da Scientifican.com.
- Prione. Recuperato da.Wikipedia.org